News
A Huntington’s disease study supporting the Neuroarcheology concept
Huntington’s disease, a dominant heritable disease that manifests in mid-adulthood, is caused by expansion of a polyglutamine tract within the huntingtin protein (HTT). The mutation leading to the polyglutamine tract confers a toxic gain of function phenotype resulting in neurodegeneration that is most severe in the striatum. Despite its “late” symptomatic manifestation, there are several clinical reports of pre-symptomatic deviations in patients with Huntington’s disease showing alterations in cerebellar-striatal connectivity1 and prefrontal cortex functionality2. In their Science3 publication, the teams of Alexandra Durr and Sandrine Humbert shows that pre-symptomatic alterations in neurodevelopment due to mutations in the HTT protein are present already at 13 weeks of gestation with abnormalities in the developing cortex, defects in cell polarity and differentiation, and cell progression. The implications of this work are important since they challenge the dissociation between developmental and neurodegenerative disorders and stress the need to evaluate early pre-symptomatic signatures in search of treatments. Interestingly, this study keeps with the Neuroarcheology4 concept proposed by Yehezkel Ben-Ari more than a decade ago which suggests that neurological disorders are “born” in utero leading to a persistence of neurons endowed with immature features that are the final cause of the deleterious sequels.
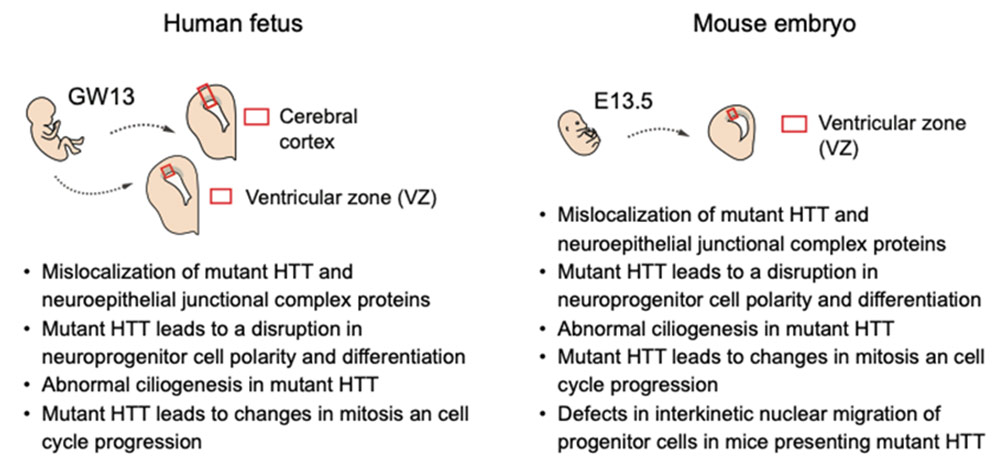
References : 1. Tereshchenko AV. et al. Abnormal development of cerebellar-striatal circuitry in Huntington disease. Neurology. 2020, 94(18):e1908-e1915. Doi: 10.1212 / WNL.0000000000009364. 2. Wolf RC. et al. Dorsolateral prefrontal cortex dysfunction in presymptomatic Huntington’s disease: evidence from event-related fMRI. Brain. 2007, 130(11):2845-57. Doi: 10.1093 / cerveau / awm210. 3. Barnat M. et al. Huntington’s disease alters human neurodevelopment. Science. 2020, 369:787-793. Doi: 10.1126/science.aax3338. 4. Ben-Ari Y. Neuro-archaeology: pre-symptomatic architecture and signature of neurological disorders. Trends in Neuroscience. 2008, 12:626-36. Doi: 10.1016/j.tins.2008.09.002

Literature Review
- Sleeping pills that cause “awakenings”? 17 February 2021
- A new report on the promising use of Bumetanide treatment for autism 24 January 2021
- A Huntington’s disease study supporting the Neuroarcheology concept 16 August 2020